









一、新冠疫情现状:全球疫苗接种水平低,病毒变异快
海外新冠疫情反复,疫苗接种率低
新冠疫情自 2020 年年中初步得到缓解后,又分别在 2020 年 10-12 月、 2021 年 4 月、2021 年 7 月经历 3 次全球大规模爆发,整体呈现出无季节性与高传播性。至今,新冠每日新增病例数量仍保持十万级水平。
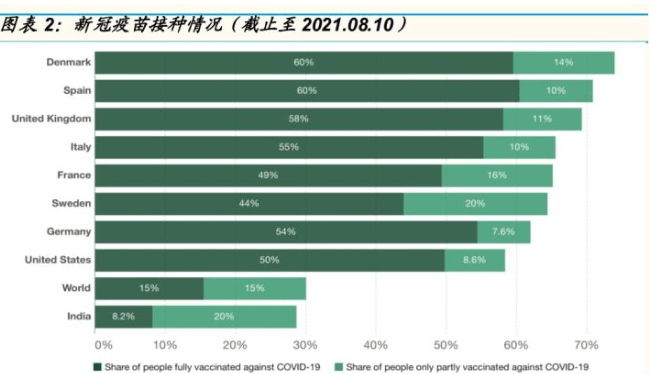
接种率低,尤以发展中国家为甚:目前,已有多款新冠疫苗可用于接种防疫。包括辉瑞的 BNT162b2, Moderna 的 mRNA-1273, 强生的 Janssen COVID-19 Vaccine 以及阿斯利康 Vaxzevria。其中,辉瑞与 Moderna 公的疫苗均基于 mRNA 技术制造。尽管多国已开展疫苗接种,世界人口整 体接种率仍处于较低水平。截止 2021 年 8 月 3 日,世界人口完全接种率14.84%,部分接种率 14.12%。疫情较为严重的发达地区(北美、欧洲) 接种率接 50%,而疫情严重的发展中国家与地区,疫苗接种率低于世界平 均水平,如非洲接种率 3.75%、印度接种率 27.62%。低接种率意味着仍 有大量人口不具备病毒免疫能力。全球短时间内仍将暴露在新冠疫情肆虐 的威胁之中。
新冠病毒变异,Delta 毒株流行,部分海外疫苗有效率下降
新冠病毒自去年起,已出现多种变异。WHO 针对新冠病毒变异情况,将主要的变异类型分为两大类:VOC (Variants of Concern) 与 VOI (Variants of Interest)。
目前,Delta 株型新冠病毒已在全球范围内成为主导,在所有分型新冠病 毒中占有绝对比例。近日国内南京爆发的新冠病例,经南京政府确认系 Delta 毒株引起。
针对 Delta 变异,疫苗的有效性出现了一定程度下降。在英国研究中使用辉瑞 BNT162b2 疫苗(2 doses),对 alpha 变体有效性为 93 %,对 delta 变体有效性为 88%;在使用阿斯利康 ChAdOx1 nCoV-19 疫苗(2 doses)时,对 Alpha 变体有效性为 66%,对 delta 变体有效性为 60%。
除 Delta 变异之外,Gamma、Lota 与 Lambda 变体在南美洲新冠分型中占比较高,其中 Gamma占比达到 50%以上,而 lota 型虽占比较低,该变体在南美呈现上升趋势。非洲地区的病毒占比情况与全球平均情况差别较大,Delta 型比重呈现下降趋势,Beta 型比重近期从降低转为升高,目前该变体占有近 30%比例。
疫苗接种率低、有效性有限,病毒持续变异,各国防疫政策差异等原因导 致疫情反复,需要构筑第三道防线即治疗药物以提升疫情控制水平。尽管现有疫苗对于各新冠病毒变体仍具有一定效力,但受到病毒持续变异的影响,有效性呈现逐步下降的趋势;另外,现阶段疫苗接种率较低、部分人群无法产生免疫应答也限制了疫苗效用;除此之外,各国防疫政策差异也 造成疫苗有效性进一步下降。诸多因素导致新冠感染人数仍高居不下,人们将面临与新冠病毒长期共存的挑战。面对新冠常态化,除疫苗免疫和常规疫情防控措施之外,治疗性药物将同样发挥重要作用。通过用药可以降低新冠住院率、致死率,最终减弱新冠对人类健康的威胁以及对公共卫生造成的巨大负担。
二、治疗性药物研发概况:中和抗体
中和抗体类药物作用机制
针对 SARS-CoV-2 的中和抗体(NAb)是用于 COVID-19 患者的一类治疗 性药物。SARS-CoV-2 病毒表面的棘突 S 蛋白(spike protein)可识别宿 主细胞受体-血管紧张素转换酶 2(ACE2)并介导膜融合,其含有的一个 C 端受体结合域(RBD)更是直接参与了宿主受体的识别,介导病毒入侵 宿主细胞。中和抗体通过特异性结合 S 蛋白,阻断病毒与宿主细胞受体 ACE2 蛋白,抑制病毒感染正常细胞。
获得紧急使用授权(EUA)的中和抗体一览
Bamlanivimab 是第一个获得 COVID-19 授权 EUA 的单克隆抗体(2020 年 11 月)。同月, FDA 还授予了再生元公司中和抗体组合疗法 REGENCOV(Casirivimab and Imdevimab)紧急使用授权。2021 年 2 月,FDA 授予了联合使用 bamlanivimab,etesevimab 用于治疗轻度至中度高危患 者的紧急使用授权。2021 年 2 月上旬,由 Celltrion 研发的 regdanvimab 中和抗体获得韩国药品安全机构的有条件上市许可。目前,Celltrion 正在 寻求欧洲药品管理局 (EMA)和 FDA 的有条件许可。最近,FDA 在 2021 年 5 月授予了 sotrovimab 用于 COVID-19 早期治疗的紧急使用授权。
礼来/君实:Bamlanivimab & Etesevimab
Bamlanivimab 是 FDA 于 2020 年 11 月 9 日授予 COVID-19 紧急使用授权的首 个单克隆抗体。bamlanivimab 和 etesevimab 联合治疗 2021 年 2 月 9FDA 批准。
作用机制:Bamlanivimab 是一种针对 SARS-CoV-2 棘突蛋白的强效、中 和性 IgG1 单克隆抗体,可以阻止病毒附着和进入人体细胞,从而中和病 毒,潜在预防和治疗 COVID-19。Etesevimab 是一款重组全人源单克隆 中和抗体,特异性结合 SARS-CoV-2 表面刺突蛋白(S 蛋白)受体结构域, 并能有效阻断病毒与宿主细胞表面受体 ACE2 的结合,由君实生物和中国科学院微生物研究所共同开发,是中国首个进入临床阶段的中和抗体药物。
临床试验进展: 2021 年 3 月 etesevimab 和 bamlanivimab 双抗体疗法发布 了 BLAZE-1 研III 期 临 床 试 验 的 新 数 据 , Etesevimab 及 Bamlanivimab 双抗体疗法明显改善 COVID-19 患者的症状并且COVID19 住院及死亡风险降低 70%。
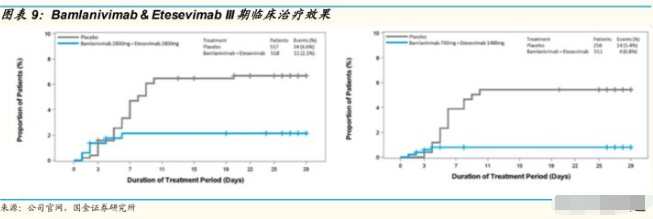
应对变异毒株效果欠佳,暂停供应 :2021 年 4 月, FDA 撤回了 bamlanivimab 单独使用授权,只被授权与 etesevimab 联用。6 月,发现 bamlanivimab 和 etesevimab 对 Beta 和 Gamma 变异株的中和能力有限, 因此双抗体疗法在美国暂停供应。bamlanivimab/etesevimab 双抗体疗法针对 Alpha 及 Delta 均有效。
商业化进展:2020 年 5 月,礼来从君实生物引进 etesevimab,获得大中华区以外地区产品临床开发、生产和商业化的独占许可。礼来向君实生物 支付 1000 万美元首付款,并在每一个君实新冠抗体(单用或组合)实现规定 的里程碑事件后,向公司支付最高 2.45 亿美元的里程碑款,外加该产品 20%的销售分成。2021 年 2 月 26 日,美国政府已决定采购至少 10 万剂, 协议金额为 2.1 亿美元,相关用药将在 2021 年 3 月 31日之前交付。
再生元/罗氏:REGEN-COV2
再生元的 COVID-19 治疗是两种单克隆抗体 casirivimab 和 imdevimab 组合, 于 2020 年 11 月 21 日获得了 FDA 的紧急授权。
作用机制:REGEN-COV2 是由 2 种单抗(casirivimab 和 imdevimab)组 成的一种鸡尾酒疗法,2 种单抗分别针对新型冠状病毒 S 蛋白受体结合区域的 2 个独立的、不重叠的位点,具有协同作用,可降低病毒变异逃逸的风险,并保护人群免受 S 蛋白发生突变的病毒变体的侵害。
临床试验进展:III 期数据优异,研究达到了主要终点:与安慰剂相比, REGEN-COV 治疗将住院或死亡风险显著降低了 70%(1200mg,静脉输 注[IV])和 71%(2400mg IV)。该研究中,REGEN-COV 也达到了全部次 要终点,包括将症状持续时间显著缩短 4 天。对直至第 169 天的所有可用 患者数据进行了安全性评估,未发现新的安全信号。严重不良事件(SAE) 主要与 COVID-19 有关,发生率 1200mg 组为 1.1%、2400mg 组为 1.3%、 安慰剂组为 4.0%。1200mg 组(n=827)有 1 例死亡,2400mg 组 (n=1849)有 1 例死亡,安慰剂组(n=1843)有 5 例死亡。6 月初, FDA 发布了一份修订后的授权,允许该公司在低剂量仍有效的情况下,将 剂量降低到原来授权剂量的一半左右,并授权在静脉注射不实用的情况下 进行皮下注射。
商业化进展:到目前为止,REGEN-COV 对所有已知的 COVID-19 变种都有效,该公司正在考虑扩大其 EUA,让未接种疫苗的高风险人员也能用于预防。和美国达成两项协议,对美国整体潜在供给量超过 150 万剂。与罗氏合作在全球制造和分销,并确保在低收入和中等收入国家的可用性。
GSK/Vir :Sotrovimab
2021 年 5 月 26 日,FDA 授予 GSK 和 Vir 的 sotrovimab 用于治疗轻度至中度 COVID-19 成人和儿科患者(12 岁及以上,体重至少 40 公斤)。
作用机制:sotrovimab 是一种具有双重作用的单克隆抗体,同时具有阻断病毒进入健康细胞、清除受感染细胞的潜力。sotrovimab 能够与 SARSCoV-2 和 SARS-CoV-1(引起 SARS 的病毒)共有的一个表位结合,该表位高度保守,可使抗药性的产生更加困难。sotrovimab 融入了 Xencor 公司的 Xtend 技术,用于在肺部实现高浓度,以确保最佳穿透受 SARSCoV-2 影响的气道组织,并具有延长的半衰期。
临床进展:今年 6 月份,Vir 公司和葛兰素史克联合宣布单抗 sotrovimab 3 期 COMET-ICH 研究最终数据 ,对全部 1057 名患者的主要疗效分析表明, 该研究达到了主要终点:到第 29 天,与安慰剂相比,sotrovimab 将住院超过 24 小时或全因死亡风险显著降低 79%(调整后的相对风险降低; p<0.001)。到第 29 天,因任何疾病的急性处理而住院超过 24 小时或任何 原因导致死亡的患者中,sotrovimab 组有 6 例(1%),安慰剂组有 30 例 (6%),疗效显著。同时,sotrovimab 对新冠病毒 变异毒株 B.1.1.7(α/英国)、B.1.351(β/南非)、P.1(γ/巴西)、 B.1.617(δ/印度)、B.1.427/B.1.429(Epsilon/加利福尼亚)和 B.1.526 (Iota/纽约)均有疗效。
商业化推进:目前,GSI 与 Vir 正积极与全球各地的政府机构合作,向需要治疗的患者提供 sotrovimab。双方计划在 2021 年下半年向美国 FDA 提交一份生物制品许可申请(BLA)。欧洲药品管理局(EMA)已经开始对 sotrovimab 数据进行滚动审查,直到有足够的证据支持提交正式的上市许 可申请为止。葛兰素史克和 Vir Biotechnology 8 月 5 日宣布,已与欧盟委员会(EU)签署联合采购协议,将提供多达 22 万剂新冠肺炎治疗药物 sotrovimab。双方的战略制造网络使其能够在美国 EUA 后的第一年制造大 约 200 万剂以支持紧急供应,现有大约 45 万剂。
Celltrion: regdanvimab
作用机制:全人源抗 SARS-CoV-2 单克隆抗体,与病毒棘突蛋白的 RBD 结合,以抑制其与受体 ACE2 的相互作用,阻断病毒的进入。此外, regdanvimab 已被证明能中和 D614G 变体,D614G 变体是最具传染性的 S 蛋白突变之一。
临床进展: 2021 年 6 月,III 期临床试验公布:与安慰剂相比, regdanvimab 将进展为重症 COVID-19 的高危患者最长至第 28 天的住院 或死亡风险显著降低了 72%,达到主要有效性终点。regdanvimab 同时显 著降低所有患者的住院或死亡风险,降幅为 70%,达到第一个关键次要终 点。同时达到其他关键的次要终点,包括症状持续时间缩短更快、更持久。
新冠病毒变异,部分中和抗体有效性降低
由于新冠病毒变异主要出现在 S 蛋白中,现有的中和蛋白将面临有效性降低的风险。由于 S 蛋白的第 501 位氨基酸能直接影响病毒与人体细胞的结合,该变异导致的最直接结果就是病毒传染性显著变强。此次全球流行 的 Delta 变异株相较于其他变异株,则在 S 蛋白上新增了 3 个重要突变: L452R、T478K 和 P681R。其中,L452R 突变既增加了 S 蛋白对受体的 亲和力,又降低了抗体识别,包括恢复期血清中存在的抗体以及一些临床上重要的中和单克隆抗体的识别;T478K 突变可能会直接增强 S 蛋白和受 体的相互作用,并以此逃避免疫系统监视;P681R 突变可间接增强 S 蛋白 介导的病毒入侵细胞过程,从而增加病毒的传染力。
中和抗体在研管线一览
除已获批的几款抗体之外,仍有大量中和抗体分子处于在研阶段。我国药企腾盛博药自研的 BRII198 & BRII196 鸡尾酒联合疗法已经入 ACTIV-3 III 期试验,国内已进入 II 期临床。百济与 Singlomics 合作开发的中和抗体 DXP-593 已经入临床 II 期,公司公告表明现已完成约 180 名轻度至中度 COVID-19 患者的登记( NCT04551898 )。阿斯利康 III 期结果表明 AZD7442 对于暴露后尚未感染的患者或有预防性效果。此外,Adagio、 Abbvie 研发的中和抗体也正有序展开临床研究;Celltrion 对已获得韩国药 品管理局临时许可的 Regdanvimab 在进行住院患者治疗有效性评估试验。
腾盛博药:BRII-196& BRII-198
作用机制:BRII-196 和 BRII-198 针对非竞争性的新冠病毒刺突蛋白的受 体结合区 RBD 中不同抗原表位,组成联合疗法,特别应用了基因工程技 术以降低抗体介导依赖性增强作用的风险,以及延长血浆半衰期以获得更 持久的治疗效果。它们的非重叠表位结合区提供了针对 SARS-CoV-2 的高 度中和活性。目前的体外证据表明,BRII-196/BRII-198 联合疗法对病毒变 异株“α”、“β”、“γ”、 “ε”以及“δ”保持中和活性。
临床进展:美国进入 III 期临床,中国进入 II 期临床。2021 年 8 月 5 日, 公司宣布 ACTIV-2 研究已在美国、巴西、南非、墨西哥和阿根廷的研究中心完成 846 位受试者的入组工作。同时,BRII-196/BRII-198 联合疗法在中国的二期临床试验(NCT04787211)正在进行,并由钟南山院士牵头。针 对中国近期出现的由“德尔塔”变异株引起的新冠病例,腾盛博药已与中国政府机构和医院开展合作,向广州、深圳、瑞丽、昆明、南京以及扬州 提供 BRII-196/BRII-198 开展临床救治。
百济/Singlomics: DXP-593/ DXP-604
作用机制:DXP-593 和 DXP-604 是从 60 多例新冠肺炎康复者抗原 B 细胞中利用高通量单细胞转录组和 VDJ 测序技术筛选出的两款新冠病毒中和 抗体。DXP-593 在假病毒和新冠病毒中分别表现出很强的中和活性,半抑 制浓度分别为 1.2 纳克/毫升和 15 纳克/毫升。在感染新冠病毒的啮齿模型中,DXP-593 也具有很强的疗效和预防效果。DXP-604 的结合表位不同 于 DXP593,也具有高中和活性。
临床进展:2020 年 9 月在澳大利亚启动关于 DXP-593 的随机、双盲、安 慰剂,对照 I 期临床试验,并于 2020 年 10 月于多个国家启动 I/II 期临床 试验。
三、治疗性药物研发概况:口服新冠药物
口服新冠药物概况
不同于中和抗体对病毒表面蛋白进行阻断,在研口服新冠药物侧重于抑制病毒转染过程(胞内为主)。因此,从用药机制分析,相对于中和抗体,口服新冠药物或对病毒变异敏感性较低。此外,口服新冠药物相对于输注给药,在用药便利性、用药成本上都有显著优势。
开拓药业:雄激素受体(AR)拮抗剂普克鲁胺
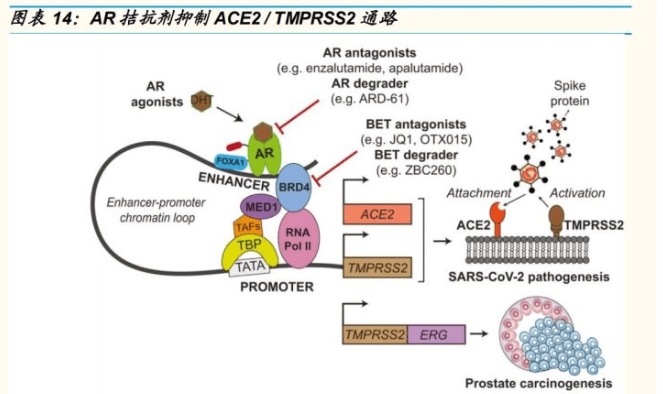
作用机制:在过去的 2020 年,研究逐步证明通过 AR 拮抗可以有效抑制新冠病毒入侵宿主的两个关键蛋白 ACE2 / TMPRSS2 通路。依此,开拓药业积极推进第二代非甾体类 AR 受体拮抗剂“普克鲁胺”用于新冠患者的临床试验。
临床试验进展:最新公布的普克鲁胺用于感染新冠患者治疗的多项临床研究数据显示出其对新冠药物的积极治疗作用。试验数据显示,该药物对于不同程度患者都具有疗效,意味着普克鲁胺或能够在治疗新冠肺炎的不同 阶段发挥作用。
基于普克鲁胺良好的临床试验数据,FDA 已批准普克鲁胺在美进行治疗轻 中症新冠男性患者的 III 期关键性全球多中心临床试验、普克鲁胺治疗住院 新冠患者的 III 期关键性临床试验。其中,NCT04869228 试验对于公司向 美国等地区申请药物 EUA 非常关键。根据公开信息披露,该试验目标入组 724 名轻中度 COVID-19 患者,试验设臵的主要终点为“在第 28 天前需氧 的受试者百分比”。预计该项试验中期结果将于今年 9 月公布。
商业进展:基于普克鲁胺良好的 III 期试验表现,开拓药业正在全力展开普 克鲁胺上市以及全球销售等商业化工作。根据公司公告,开拓药业与复兴 医药产业就普克鲁胺治疗新冠在印度和 28 个非洲国家的商业化达成合作 协议,双方相互合作、共同推进普克鲁胺新冠适应症的紧急使用授权(EUA) 申请、推广和销售工作。2021 年 7 月 16 日公司公告称,巴拉圭国家公共 卫生和社会福利部(MSPBS)于近期正式授予普克鲁胺紧急使用授权(EUA), 用于新冠住院患者的治疗。巴拉圭 MSPBS 批准普克鲁胺 EUA 许可后,隶属于巴拉圭 MSPBS 医院系统的 Barrio Obrero 是首家使用普克鲁胺治疗新 冠患者的医院。
默沙东/ Ridgeback:Molnupiravir(MK-4482/EIDD-2801)
作用机制:Molnupiravir 是 EIDD-1931 的异丙酯前药,进入体内后水解为原型药物 EIDD-1931。目前研究认为,该药物分子通过诱导病毒 RNA 变异以至病毒 无法复制来抑制新冠病毒感染正常细胞,研究数据也证明其体内药效和 MERS-CoV 基因组 RNA 的突变率相关。体外抗病毒活性测试已证实 Molnupiravir 在几种 SARS-CoV-2 模型中具有活性,或可用于预防、治疗 COVID-19。
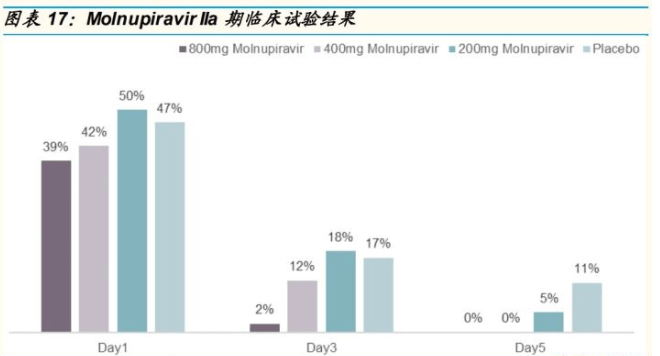
临床试验进展:2021 年 4 月 15 日,默沙东和 Ridgeback 提供了 Molnupiravir II/III 期 试 验 “ MOVe-OUT and MOVe-IN clinical trial ” (NCT04575584)最新进展。基于(i)该试验 2 期的中期分析;(ii)两项正在 进行的 II/III 期安慰剂对照药物剂量发现试验(方法:每天给药两次,持续 给药五天,评估了门诊患者(MOVe-OUT)和住院患者(MOVe-IN))(iii) 之前在门诊完成的 2a 期剂量范围研究,公司决定对 COVID-19 门诊患者 (MOVe-OUT)进行该试验 III期工作,即对门诊患者评估一天两次给药, 每次 800 mg Molnupiravir 效果。同时, 基于已有结果 ,公司表明 Molnupiravir 或许对于住院患者无效,因此,已经暂停对住院患者使用 Molnupiravir 的 III期临床计划。
商业进展:Molnupiravir 在商业落地方面推进迅速。2021 年 6 月 9 日,默 沙东发布声明称,与美国政府达成了 Molnupiravir(MK-4482)的采购协 议。协议中美国政府承诺在 Molnupiravir 获 FDA 批准用于 EUA 或获 FDA 批准上市后,将采购 Molnupiravir 约 170 万个疗程。同时,为了促进该药 物在中低等收入国家(LMIC)早日获得 EUA 授权或上市许可,默沙东在 今年 3-4 月陆续与印度当地多家医药制造公司签订了非独家无偿许可协议。 2021 年 6 月 29 日,其中 5 家公司宣布将合作开展研究性口服抗病毒药物 Molnupiravir 的临床试验,以治疗印度门诊环境中的轻度 COVID-19 患者。
罗氏/ Atea:AT-527
作用机制:AT-527 由罗氏与 Atea Pharmaceuticals, Inc. 共同开发,该药 物分子是一种研究性的口服嘌呤核苷酸前药,通过独特地抑制病毒 RNA 依赖性 RNA 聚合酶发挥作用。
临床试验进展:该项中期分析纳入了 70 名住院、高危 COVID-19 患者, 其中 62 名患者的数据可用于病毒学分析。病毒学结果表明 AT-527 迅速降 低了病毒载量水平。 在第 2 天,与安慰剂相比,接受 AT-527 的患者比基 线病毒载量平均降低了 80%,这一指标,即病毒载量的差异一直持续到第 8 天。
罗氏正在积极筹备多项评估 AT-527 用于治疗 COVID19 患者安全性、有效性的试验。从试验设臵上看,研发厂商试图更进一步 探究 AT-527 在不同程度 COVID-19 患者的有效性以及该药物对群体暴露 于 COVID-19 后进行预防性治疗的潜力,以提升产品差异。
辉瑞:口服蛋白酶抑制剂 PF-07321332
作用机制:在 SARS-CoV-2 进入细胞释放出病毒 RNA 感染正常细胞的过 程中,需要携带一种类似 3C 的蛋白酶(3CLpro),3CLpro 蛋白酶在多种冠 状病毒的生命周期中起到重要作用。辉瑞公司开发的口服 COVID-19 药物 PF-07321332 正是一款 3CL 蛋白酶的抑制剂,通过靶定 3CL 蛋白酶阻断 病毒复制。
临床试验进展:在健康志愿者的 1 期药代动力学研究中,PF-07321332 显示出超过 10 天的高药物暴露活性, 超过预期抑制 SARS-CoV-2 病毒复制的暴露水平五倍更多。基于这些数据, 辉瑞于 2021 年 7 月启动了一项针对 COVID-19 患者的 2/3 期试验。该试 验的数据预计将于 2021 年第四季度发布。
在研口服新冠药物总结
与现有治疗药物互补:不同于中和抗体对病毒表面蛋白进行阻断,在研口 服新冠药物侧重于抑制病毒转染过程(胞内为主)。因此,从用药机制分析, 相对于中和抗体,口服新冠药物或对病毒变异敏感性较低。此外,口服新 冠药物相对于输注给药,在用药便利性、药物成本上都有显著优势。
临床进展对比:4 种口服新冠药物目前均已进入临床三期,预计 均在 2021Q3-Q4 公布关键试验数据,若临床试验结果理想,进展快的产 品最早有望在 2021 年底前在欧美上市。
口服新冠药物横向对比:基于已有的试验结果和各公司接下来的试验计划, 我们可以从不同维度分析、推测药物未来应用情况。从用量上看,现阶段 在研口服新冠药物并无太大差异,对于患者依从性影响较小。而各在研药 物在治疗潜力、覆盖区域维度存在一定差异化,这将一定程度影响各公司 未来的市场空间和推广策略。
四、关注新冠相关 CDMO 放量潜力
疫苗及中和抗体 CDMO
新冠疫情后,相关疫苗和中和抗体等项目快速立项、推进,与之相关的研 发、开发和生产需求激增。全球来看,高效、优质、稳定的研发、开发和 生产大分子的产能相对稀缺,LONZA\CATALENT\药明生物相关订单需求 快速增长。目前疫情后的第一阶段,疫苗产能逐渐释放,相关大分子 CDMO 企业的疫苗类订单及业绩快速增长;我们未来 12-24 个月,随着新 型疫苗和中和抗体管线的逐渐向后推进和获批放量,新冠相关大分子 CDMO 订单仍有望进一步增长。
药明生物在疫情期间爆发了强大的紧急事件应变能力和执行力,高效地推 进新冠疫苗及中和抗体项目,并成功将新冠中和抗体的 DNA 到 IND 交付间缩短到 3-5 个月,成功赋能了 2 个新冠疫苗和 12 个新冠中和抗体项目。
小分子 CDMO
小分子药物的生产过程大致可以分成以下几个阶段:偏精细化工阶段 (nonGMP)→RSM(nonGMP)→GMP 中间体、高级中间体→API (GMP)→制剂(GMP)。过去 15-20 年时间,国内头部的 CDMO 企业 不断实现国际医药创新高端制造供应链上的突破,国际认可度持续提升, D(Development)和 M(Manufacture)的产品不断升级(RSM→GMP 中间体→GMP 高级中间体→API)。参考我们对口服新冠药物稳态市场规 模的分析和小分子创新药制造产业链的价值链切分,我们预计完整的口服 新冠药物制造环节空间较大,中国境内口服新冠药物的高级中间体、原料 药 CDMO 订单有望快速放量。
五、风险提示
新药研发失败风险;疫情等因素导致新药临床开发进度低于预期风险;行业及竞争加剧导致原有产品放量不及预期风险;监管风险;核心技术人员流失风险;部分原材料海外供应风险等。










